
Разработка стратегии регистрации по требованиям ЕАЭС
Регистрация лекарственного препарата (ЛП) – сложный, комплексный процесс, требующий соблюдения большого количества требований и норм. Основные принципы этого процесса отражены в документах Евразийской экономической комиссии. Зная все регуляторные вопросы и нюансы процедур регистрации ЛП, можно разработать правильную и эффективную стратегию, без которой невозможен процесс получения регистрационного удостоверения (РУ).
Основной документ, которым следует руководствоваться при разработке стратегии − Решение Совета ЕЭК №78 от 03.11.2016 «О Правилах регистрации и экспертизы лекарственных средств для медицинского применения». Он устанавливает принципы, порядок, сроки регистрации и экспертизы лекарственных препаратов, а также условия обращения ЛП в государствах-членах ЕАЭС. И включает информацию о требованиях к регистрационному досье: описание модулей, формы заявлений, отчетов и прочих документов, необходимых для регистрации ЛП.
Основные этапы, которые имеют наиболее важное стратегическое значение в процессе подготовки регистрационного досье.
- Описание регистрируемого препарата.
При разработке стратегии следует начать с определения регистрационной группы препарата. В соответствии с Приложением №1 к Правилам регистрации препарат можно отнести к группе оригинальных, воспроизведенных, гибридных, биоаналагов или иным группам. От классификации зависит формат подаваемого досье и выстраивание всей последующей стратегии.
- Выбор механизма регистрации.
Существуют два механизма осуществления государственной регистрации в рамках единого рынка ЕАЭС.
- Процедура взаимного признания. Регистрация осуществляется только в одном выбранном государстве-члене ЕАЭС (референтном государстве), с возможностью дальнейшего признания в иных государствах-членах ЕАЭС по желанию заявителя.
- Децентрализованная процедура. Регистрация осуществляется одновременно несколькими государствами-членами, с выбором референтного государства.
- Определение объема данных в досье.
Для получения РУ необходимо предоставить информацию обо всех жизненных циклах препарата: научном изыскании, доклинической разработке, клинической разработке, фармацевтической разработке и процессах производства. В регистрационное досье в соответствии с требованиями ЕАЭС, следует включить информацию:
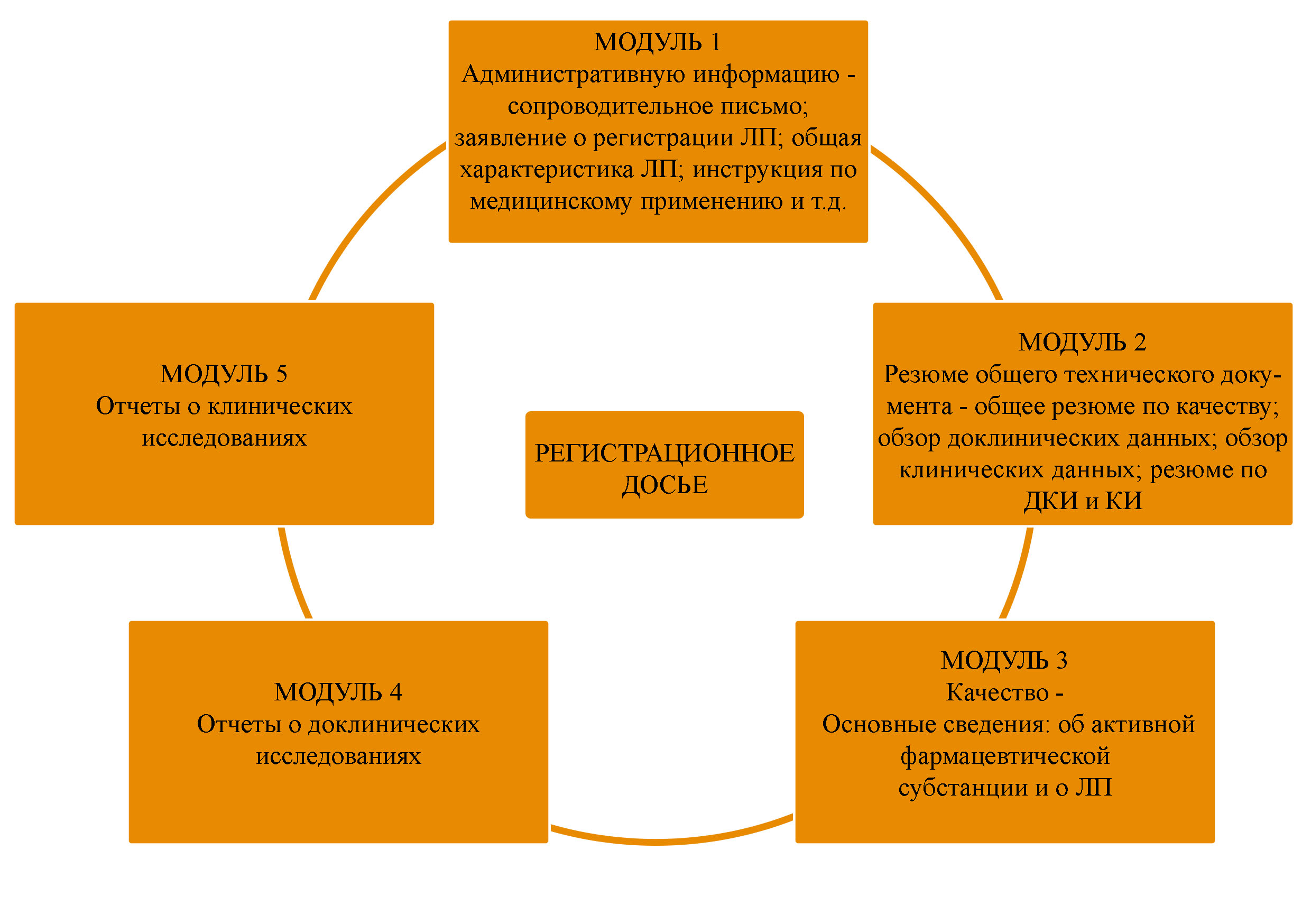
4. Проведение исследований.
Следующий этап — разработка плана проводимых доклинических и клинических исследований. Актуальная документация, в которой описаны требования к проведению клинических исследованиях находится на сайте Евразийской Экономической комиссии.
Грамотно составленная стратегия позволит: составить план действий, определить сроки всех необходимых этапов при подготовке регистрационного досье, сэкономить средства, и успешно, с минимальными затратами, пройти регистрацию лекарственного препарата.
Наши специалисты обладают значительным опытом работы в данной сфере, и будут рады помочь в разработке стратегии регистрации для вашего препарата.
